
Anno X, Numero 3 - ottobre 2016
- Malattia di Pompe a insorgenza tardiva: conoscerla, riconoscerla
La malattia di Pompe è una patologia del metabolismo del glicogeno, ereditaria e potenzialmente fatale, dovuta a un deficit dell'attività dell'?-glucosidasi acida (GAA) lisosomiale; il deficit enzimatico comporta un accumulo di glicogeno intralisosomiale in particolare nel tessuto muscolare, con un danno muscolare irreversibile.1,2
La malattia di Pompe, nota anche come deficit di maltasi acida (acid maltase deficiency, AMD) o glicogenosi di tipo II (glycogen storage disease type II, GSDII),3 è una malattia genetica a trasmissione autosomica recessiva causata da una mutazione del gene sul braccio lungo del cromosoma 17; si può manifestare quando entrambi i genitori sono portatori del gene mutato (Fig. 1).
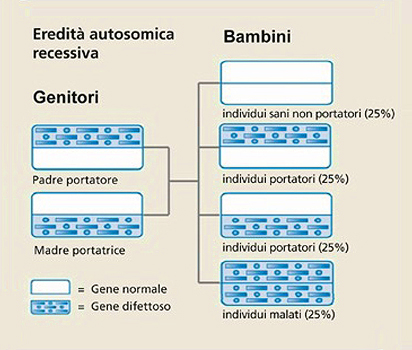
Figura 1. Ereditarietà e malattia di Pompe
La malattia ha un ampio spettro di fenotipi clinici; viene classificata comunemente in una forma infantile, con esordio nei primi tre-sei mesi di vita, e in una forma tardiva (late onset Pompe disease, LOPD) che può insorgere in età infantile, giovanile o adulta, ma si presenta comunque dopo il primo anno di età.1 La progressione della malattia si differenzia in base alla forma clinica e all'età di esordio. La forma infantile è caratterizzata da cardiomiopatia ed exitus solitamente entro il primo anno di vita; nella forma tardiva la progressione, più lenta ma comunque inesorabile, causa morbilità significative e ha un impatto sfavorevole sulla qualità della vita (Fig. 2), portando spesso alla necessità di dover ricorrere alla sedia a rotelle e/o a supporti per la ventilazione.
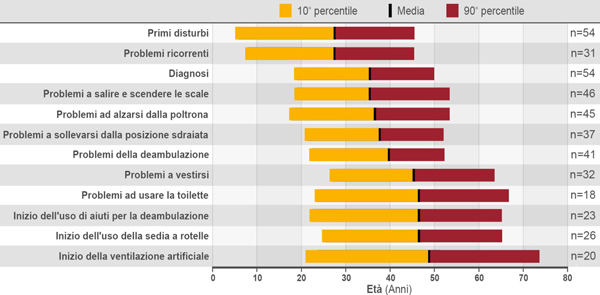
Figura 2. Distribuzione dell'età in relazione ad eventi specifici durante la malattia di Pompe4mod
L'aspettativa di vita nei pazienti affetti dalla forma tardiva è variabile; il coinvolgimento cardiaco è raro, mentre la morte prematura, in genere a causa di insufficienza respiratoria, può essere un evento comune.
Una diagnosi precoce e il trattamento della malattia di Pompe possono portare a un miglioramento della qualità ed aspettativa di vita dei pazienti con questa malattia neuromuscolare progressivamente invalidante.1,5
Segni e sintomi
- aumento dei livelli di creatinchinasi
- progressiva debolezza della muscolatura prossimale
- insufficienza respiratoria.
1. Aumento dei livelli di creatinchinasi
Alcuni studi, che hanno valutato l'iperCKemia come segno potenzialmente indicativo di LOPD, hanno evidenziato che:
- su 207 pazienti con iperCKemia, il 2,4% aveva la malattia di Pompe7
- su 137 pazienti con iperCKemia paucisintomatica, la prevalenza di malattia di Pompe a esordio tardivo era del 2,2%8
- su 104 pazienti con iperCKemia asintomatica o minimamente sintomatica, 4 casi (3,8%) si correlavano a un deficit di GAA.9
Tra i test di laboratorio da prescrivere in caso di sospetto diagnostico di malattia di Pompe deve quindi essere previsto il dosaggio della CK,2 che rappresenta un primo step semplice e relativamente poco costoso.
In conclusione, la progressiva debolezza muscolare prossimale, associata o meno a insufficienza respiratoria o iperCKemia, può far sospettare la malattia di Pompe.3,10
I pazienti con sintomi di debolezza muscolare prossimale dei cingoli e i pazienti con iperCKemia persistente devono pertanto essere sottoposti a screening per la malattia di Pompe.1,10,11
2. Debolezza muscolare
?? il quadro clinico più comune; i muscoli degli arti, in particolare degli arti inferiori, e del tronco sono interessati precocemente nel corso della malattia (Fig. 3).12
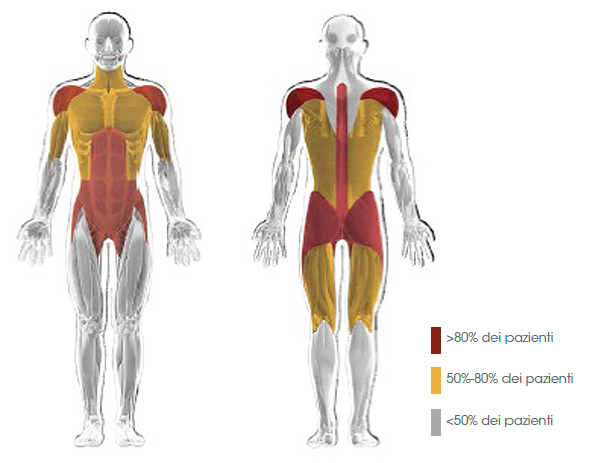
Figura 3. Distribuzione della debolezza muscolare prossimale tipica dei pazienti con malattia di Pompe a esordio tardivo12mod
L'ipostenia, più prossimale che distale, colpisce prevalentemente i muscoli del dorso e del cingoli pelvico e scapolare,2 con la comparsa di una tipica andatura "anserina". I pazienti possono manifestare dolori muscolari, essere soggetti a frequenti cadute e lamentare facile affaticabilità. Nei pazienti con malattia di Pompe a esordio tardivo, l'elettromiografia (EMG) spesso può mostrare alterazioni a livello dei muscoli paraspinali, sotto forma di scariche miotoniche (Fig. 4).1

Figura 4. Quadro elettromiografico di paziente con malattia di Pompe
- il 15% di 132 pazienti con debolezza prossimale cingolare è risultato positivo al dried blood spot test (screening di primo livello per la malattia di Pompe)11
- su 144 pazienti con miopatia cingolare o debolezza muscolare prossimale non altrimenti diagnosticata, il 10,2% aveva la malattia di Pompe7
- su 38 pazienti con distrofia muscolare cingolare non classificata, circa l'8% aveva una malattia di Pompe a esordio tardivo.13
3. Insufficienza respiratoria
In un terzo circa dei casi i soggetti affetti da LOPD possono andare incontro a una compromissione della funzionalità respiratoria; questa è conseguente a una riduzione progressiva dei flussi e volumi polmonari causata dalla debolezza del diaframma e dei muscoli respiratori accessori. Si può quindi assistere alla comparsa di un quadro di insufficienza respiratoria di tipo restrittivo, con sintomi consistenti in cefalea notturna o al risveglio, sonnolenza, ortopnea e dispnea dopo aver svolto attività fisica.
La diagnosi
La valutazione clinica del paziente con sospetta malattia di Pompe deve includere, oltre alla dimostrazione della ridotta attività enzimatica mediante test biochimico, la biopsia muscolare; tuttavia, meno dell'80% delle biopsie muscolari mostrano un anomalo contenuto di glicogeno, suggestivo di malattia di Pompe, e l'assenza di accumulo di glicogeno può dare dei falsi negativi.10 Per giungere alla conferma diagnostica è necessaria la caratterizzazione genetica mediante studio di sequenza del gene codificante l'enzima GAA.
Per quanto riguarda il test biochimico, di recente è stata introdotta la possibilità di fare diagnosi di malattia di Pompe su una goccia di sangue essiccata su filtri di carta bibula attraverso il dried blood spot (DBS) test, che si è rivelato particolarmente utile come screening di primo livello.11 Il kit diagnostico consente la raccolta, l'utilizzo e la spedizione dei campioni raccolti a un laboratorio di riferimento; in caso di positività, è comunque necessaria la conferma diagnostica biochimica e/o molecolare.1
La disponibilità di questo test ematico, rapido e affidabile, può consentire di cambiare la prospettiva per i pazienti con malattia di Pompe,11 per i quali il rischio di dover ricorrere alla sedia a rotelle o a un supporto respiratorio aumenta rispettivamente del 13 e dell'8% ogni anno successivo alla diagnosi.14
Conseguenze del ritardo diagnostico
Nella malattia di Pompe a esordio tardivo si è osservato un ritardo diagnostico di 6 anni (Fig. 5).15
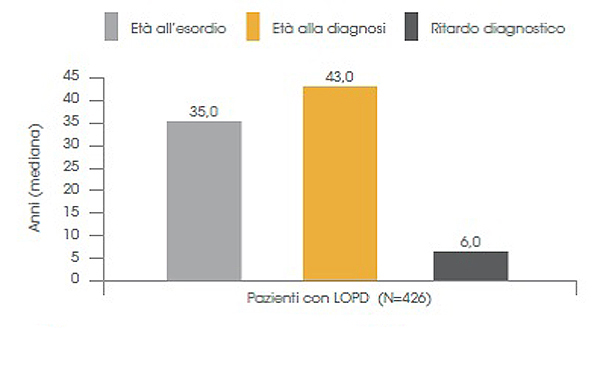
Figura 5. Ritardo diagnostico nella popolazione di pazienti con età di esordio dei sintomi >12 anni15
?? stato dimostrato che quanto più tardiva è la diagnosi, tanto maggiori sono le probabilità di dover ricorrere alla sedia a rotelle e dipendere dal ventilatore.14
Una diagnosi tempestiva è quindi di fondamentale importanza per poter iniziare la terapia il più precocemente possibile e modificare la storia naturale della malattia.
Prospettive
La disponibilità della terapia enzimatica sostitutiva con GAA ricombinante consente di migliorare le funzioni motorie e respiratorie e di prolungare in modo significativo la sopravvivenza dei pazienti affetti da LOPD. Il cardine della gestione terapeutica è rappresentato dai Centri di eccellenza dislocati sul territorio italiano, gestiti prevalentemente da specialisti neuromuscolari. Importante è anche l'appoggio offerto dalle associazioni pazienti, e in particolare dall'Associazione Italiana Glicogenosi (AIG, www.aig-aig.it/), che si adoperano per informare, rassicurare, raccordare le famiglie e tenere i contatti con le realtà scientifiche che operano nel settore della ricerca e della terapia; tra queste, l'Associazione Italiana di Miologia (AIM, www.miologia.org/) raduna i professionisti che operano nel campo delle malattie neuromuscolari presso strutture Universitarie, Ospedaliere, IRCCS ed altre strutture sanitarie al fine di promuovere e divulgare le conoscenze nel campo neuromuscolare nell'interesse dei pazienti.
A cura della Redazione
Bibliografia
- American Association of Neuromuscular & Electrodiagnostic Medicine. Diagnostic criteria for late-onset (childhood and adult) Pompe disease. Muscle Nerve 2009; 40 (1): 149-16.
- Kishnani PS et al. Pompe disease diagnosis and management guideline. Genet Med 2006; 8 (5): 267-88.
- Hirschhorn R et al. Glycogen storage disease type II: acid ?-glucosidase (acid maltase) deficiency. In: Scriver CR et al, eds. The Metabolic & Molecular Bases of Inherited Disease. 8th ed. New York, NY: McGraw-Hill; 2001: 3389-420.
- Hagemans ML et al. Clinical manifestation and natural course of late-onset Pompe's disease in 54 Dutch patients. Brain 2005; 128:671-7
- Gungor D et al. Impact of enzyme replacement therapy on survival in adults with Pompe Disease. Result from a prospective observational interntation study. Orphanet J Rare Dis 2013, 8: 49
- Kishnani PS et al. Pompe disease diagnosis and management guideline. Genet Med 2006; 8: 267-88.
- Bautista Lorite J. Detección de la enfermedad de Pompe en pacientes con distrofia de cinturas indefinidas o hiperCKemias asintomáticas. Expert Rev Neur Ed especial Octubre 2013:17-9.
- Spada M et al. Screening for later-onset Pompe's disease in patients with paucisymptomatic hyperCKemia. Mol Genet Metab 2013; 109 (2): 171-3.
- Fernandez C et al. Diagnostic evaluation of clinically normal subjects with chronic hyperCKemia. Neurology 2006; 66 (10): 1585-7.
- Vissing J et al. Diagnosis of Pompe disease: muscle biopsy vs blood-based assays. JAMA Neurol 2013;70 (7):923-7.
- Goldstein JL et al. Screening for Pompe disease using a rapid dried blood spot method: experience of a clinical diagnostic laboratory. Muscle Nerve 2009; 40 (1): 32-6.
- van der Beek N et al. Clinical features and predictors for disease natural progression in adults with Pompe disease: a nationwide prospective observational study. Orphanet J Rare Dis 2012; 7: 8.
- Preisler N et al. Late-onset Pompe disease is prevalent in unclassified limb-girdle muscular dystrophies. Mol Genet Metab 2013; 110 (3): 287-9.
- Hagemans ML et al. Disease severity in children and adults with Pompe disease related to age and disease duration. Neurology 2005; 64 (12): 2139-41.
- Kishnani PS et al. Pompe Registry Boards of Advisors. Timing of diagnosis of patients with Pompe disease: data from the Pompe Registry. Am J Med Genet A 2013; 161 (10): 2431-43.



