On Medicine
Anno IX, Numero 9 - ottobre 2015


IL PARERE DELLO SPECIALISTA
Farmacologia clinica dei farmaci anticoagulanti
Marco Moia Centro Emofilia e Trombosi - Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico di Milano

Premessa
Per quasi mezzo secolo eparina e cumarinici hanno rappresentato le uniche scelte per iniziare e condurre una terapia anticoagulante. Sono farmaci scoperti "per caso" oltre cinquant‘anni fa, complessi nel loro meccanismo d‘azione, con elevate potenzialità ed altrettanto elevati rischi. Il percorso compiuto per ottimizzarne l‘uso da parte dei ricercatori e dei medici a tutti i livelli (laboratoristi, epidemiologi, clinici) ha portato, nel tempo, a sfruttarli adeguatamente, con risultati ottimi in termini di efficacia e sufficienti in termini di sicurezza. Questo percorso di miglioramento non può ancora ritenersi concluso, così come la storia di questi farmaci. Infatti, nonostante l‘enorme sviluppo della ricerca farmacologica del settore, avvenuto soprattutto nell‘ultimo decennio, eparina e cumarinici mantengono alcune indicazioni nelle quali, per il momento, non trovano validi sostituti: ad esempio, i circuiti extracorporei per l‘eparina e le protesi valvolari cardiache meccaniche per i cumarinici.
I nuovi farmaci anticoagulanti, principalmente per uso orale, hanno dimostrato elevate caratteristiche di efficacia, sicurezza e tollerabilità. Con il loro avvento si propongono nuovi scenari nella gestione delle terapie anticoagulanti, con la possibilità di espanderne l‘uso a molti pazienti che, per motivi legati alla complessità dei "vecchi" farmaci, hanno di fatto fino ad oggi ricevuto trattamenti non adeguati. Vengono di seguito presentati dunque, in modo riassuntivo, i "vecchi" (ma pur sempre indispensabili) farmaci anticoagulanti, e, successivamente, le nuove molecole. Il potenziale utilizzo dei nuovi farmaci in molte migliaia di pazienti rende necessaria una loro più approfondita conoscenza che andrà a sua volta ampliandosi e completandosi negli anni futuri, di pari passo con l‘esperienza clinica quotidiana.
Anticoagulanti parenterali
Eparina
L‘eparina, scoperta quasi un secolo fa (1), è una miscela di mucopolisaccaridi altamente solfatati, eterogenea per i diversi pesi molecolari delle varie molecole che la costituiscono (da 3000 a 30.000 kDa) che dimostrano attività anticoagulanti e farmacocinetiche diverse (2). Non più di un terzo delle molecole di eparina possiedono la sequenza pentasaccaridica che è responsabile di larga parte del suo effetto anticoagulante (3). L‘eparina agisce catalizzando l‘attività inibitoria dell‘antitrombina (AT), legandosi ai residui a carica positiva e producendo un cambiamento conformazionale nel sito reattivo argininico dell‘AT che la rende molto più rapida e attiva nella sua azione di inibizione delle proteasi seriniche. Il sito argininico dell‘eparina si lega in modo covalente alla trombina (FIIa) e ad altri enzimi della coagulazione attivati, bloccandone irreversibilmente la loro attività procoagulante. Quindi l‘eparina si dissocia dall‘AT e può attivarne un‘altra molecola. Il complesso eparina-AT inattiva i fattori della coagulazione IIa, Xa, IXa, XIa e XIIa. Le molecole di eparina con catene più corte (<18 unità saccaridiche) non sono in grado di legare la trombina, ma solo il FXa (4). Questa caratteristica è stata sfruttata dalla ricerca farmacologica nella realizzazione delle eparine a basso peso molecolare e, successivamente, del pentasaccaride sintetico fondaparinux.

L‘eparina, al pari dei suoi derivati, non è assorbita per via orale. La somministrazione è pertanto per via endovenosa (e.v.), preferibilmente per infusione continua, o sottocutanea (s.c.). Tuttavia, a causa della ridotta biodisponibilità dell‘eparina quando iniettata s.c., se si utilizza tale via di somministrazione, le dosi di eparina devono essere di regola più elevate rispetto a quelle programmate per via e.v. (5). Una volta entrata in circolo, l‘eparina si lega a numerose proteine plasmatiche, oltre che all‘AT, e questo determina una riduzione della sua attività anticoagulante (6). L‘eparina è eliminata da un meccanismo rapido saturabile (principalmente il legame con cellule e proteine) e da uno molto più lento, in larga parte renale. Alle dosi terapeutiche la maggior parte dell‘eparina è eliminata dal meccanismo rapido saturabile dose-dipendente (7,8). La complessa cinetica di eliminazione rende la risposta anticoagulante non lineare a dosi terapeutiche, con intensità e durata dell‘effetto che aumentano in modo non proporzionale all‘aumento della dose. Ad esempio, l‘apparente emivita dell‘eparina aumenta approssimativamente dai circa 30 minuti per un bolo e.v. di 25 U/kg, ai circa 60 minuti per un bolo di 100 U/kg, ai circa 150 minuti per bolo di 400 U/kg (9). L‘effetto anticoagulante dell‘eparina per via e.v. viene generalmente monitorato con il tempo di tromboplastina parziale attivato (aPTT) (10). Il tempo di coagulazione attivato (ACT) è utilizzato quando vengono impiegate dosi più alte di eparina, tipicamente nei circuiti extracorporei, in quanto risulta scarsamente sensibile alle dosi terapeutiche. Purtroppo, nonostante l‘eparina sia in uso da moltissimi anni, gli intervalli terapeutici suggeriti (prolungamento dell‘aPTT da 1.5 a 2.5 volte) sono stati suggeriti in base a studi retrospettivi. Inoltre, non è mai stata attuata una vera standardizzazione dei risultati di laboratorio in base alla sensibilità dei reagenti impiegati per la determinazione dell‘aPTT, che possono avere sensibilità assai diverse e quindi fornire risultati non confrontabili (11).
Limitazioni dell‘eparina
Le eparine a basso peso molecolare (EBPM) sono state ricavate da depolimerizzazione chimica o enzimatica dell‘eparina. Il peso molecolare medio delle EBPM attualmente in commercio è generalmente compreso tra 4000 e 5000 kDa (che corrisponde mediamente a circa 15 unità saccaridiche), con un range fra 2000 e 9000 kDa (14). Le EBPM differiscono nel meccanismo d‘azione dall‘ENF per un rapporto di inattivazione fra FXa e FIIa nettamente più elevato. Infatti, le molecole a più basso peso molecolare presenti nelle EBPM non permettono all‘AT di legare anche il FIIa, legame per il quale sono necessarie almeno 18 unità saccaridiche e che risulta indispensabile per la sua inattivazione.
Le varie molecole di EBPM presenti sul mercato differiscono l‘una dall‘altra per metodo di preparazione, peso molecolare medio e distribuzione. Queste differenze condizionano anche l‘emivita delle varie molecole che, di fatto, risulta diversa. Pertanto, non è possibile stabilire una equivalenza nelle dosi delle diverse molecole, che vanno scelte in base alle raccomandazioni di ogni singola molecola riportate in scheda tecnica.
Le EBPM vengono somministrate a dosi fisse: tali dosi vengono aggiustate sul peso corporeo del paziente per i regimi terapeutici. Di regola non viene attuato alcun monitoraggio né, conseguentemente, alcun aggiustamento della dose. Un dosaggio dell‘attività anti-FXa è stato tuttavia proposto in alcune situazioni particolarmente impegnative (gravidanza, obesità, insufficienza renale).

Farmacocinetica
Le EBPM hanno significativi vantaggi farmacocinetici rispetto all‘ENF in quanto dimostrano una più alta biodisponibilità dopo somministrazione s.c. (circa 90%) ed un effetto anticoagulante più prevedibile. Con somministrazione s.c., il picco di attività anti-FXa è tra le 3 e le 5 ore, e l‘emivita tra le 3 e le 6 ore (15). L‘eliminazione delle EBPM è prevalentemente renale (16), fatto che determina difficoltà d‘uso nei pazienti con insufficienza renale: generalmente le EBPM a dosaggio terapeutico vengono controindicate in pazienti con CrCl ≤30 ml/min. Rispetto all‘ENF, le EBPM determinano un minor rischio di HIT e di osteoporosi. Tuttavia, nei pazienti che abbiano già sviluppato HIT, esiste cross-reattività tra EBPM ed anticorpi anti-PF4 che ne controindica l‘uso.
Antidoto
Empiricamente si suggerisce che 1 mg di solfato di protamina possa neutralizzare circa 100 U anti-FXa di EBPM (17).
Fondaparinux
Il pentasaccaride sintetico ad alta affinità per l‘antitrombina fondaparinux è stato isolato agli inizi degli anni ‘80 (18). Ha un peso molecolare di 1728 kDa con una attività specifica anti-FXa di circa 7 volte superiore a quella delle EBPM (circa 700 U/mg). La sua emivita, dopo somministrazione s.c., è di circa 17 ore nel soggetto giovane, e si prolunga fino a circa 21 ore nell‘anziano. L‘effetto anticoagulante è prevedibile con una farmacocinetica lineare per dosi comprese tra 2 mg ed 8 mg s.c., o tra 2 mg e 20 mg e.v. (19). Il legame di fondaparinux con le proteine plasmatiche è minimo ed aspecifico, fatta eccezione, ovviamente, per il forte legame con AT.
Fondaparinux agisce determinando un cambiamento conformazionale del sito attivo dell‘antitrombina, che aumenta la sua reattività esclusivamente verso il FXa (Fig. 1) (20). Quindi, fondaparinux viene rilasciato dall‘AT ed è disponibile per attivare altre molecole di AT. Viene escreto immodificato con le urine ed è controindicato nei pazienti con grave insufficienza renale (CrCl <30 ml/min) per accumulo con aumento del rischio emorragico.
Le dosi di fondaparinux nella terapia del tromboembolismo venoso sono stabilite in base al peso corporeo del paziente e non vengono monitorate con test di laboratorio. Non esiste un antidoto specifico per fondaparinux: il solfato di protamina risulta inefficace. Alcuni dati indicano la potenziale utilità del FVIIa ricombinante (21), con tutti i limiti determinati dal costo e dalla potenziale trombogenicità di tale farmaco. Gli effetti collaterali non emorragici di fondaparinux sono rari e, sebbene siano stati riportati isolati casi di HIT associati a fondaparinux, vi sono ampie casistiche di pazienti con HIT trattati con successo con fondaparinux (22). Vi sono scarsi dati sulla sicurezza di fondaparinux in gravidanza: tuttavia, il passaggio transplacentare sembra minimale (23).
Inibitori diretti della trombina
Sono farmaci che non richiedono un cofattore plasmatico per esercitare l‘attività inibente l‘enzima target, vale a dire il FIIa (trombina) (Fig. 1). Il primo di tali farmaci a rendersi disponibile per l‘uso clinico è stato lepirudina, un polipeptide ricombinante di 65 aminoacidi, originariamente isolato dalla saliva della sanguisuga (Hirudo medicinalis) (24). Già approvata per l‘uso nella terapia della HIT in Europa, recentemente ne è stata cessata la produzione e non è pertanto più disponibile. Sono invece disponibili in Europa per uso clinico due diverse molecole di inibitori diretti del FIIa: bivalirudina e argatroban. Non esistono attualmente antidoti specifici degli inibitori diretti parenterali della trombina che, tuttavia, per la loro breve emivita non richiedono quasi mai di essere inattivati. L‘emodialisi e l‘emofiltrazione rimuovono, comunque, bivalirudina e argatroban dal circolo.

Bivalirudina
Peptide sintetico di 20 aminoacidi analogo dell‘irudina, si lega al sito attivo della trombina formando un complesso stechiometrico 1:1 (25). Una volta legatosi, tuttavia, perde rapidamente la sua attività. L‘emivita di bivalirudina è 25 minuti dopo iniezione e.v.; solo il 20% è escreto per via renale.
Bivalirudina è utilizzata, in alternativa all‘eparina, nei pazienti sottoposti a coronaroplastica per angina instabile, o infarto del miocardio, o in quelli con HIT candidati ad interventi coronarici (26).
Argatroban
Piccola molecola (500 kDa) che determina inibizione competitiva della trombina tramite legame non covalente che inattiva il FIIa formando un complesso reversibile (27). L‘emivita di argatroban è 45 minuti. È metabolizzato a livello epatico dal citocromo P450 3A4/5. Pertanto vi sono cautele d‘uso nel paziente epatopatico.
È approvato per l‘uso nella HIT e negli interventi coronarici percutanei ove sia controindicata l‘eparina per recente o pregressa HIT. Viene somministrato per via e.v. continua, aggiustando la dose per mantenere un aPTT ratio tra 1.5 e 2.5 (28).
Farmaci anticoagulanti orali
Cumarinici
Meccanismo d‘azione
I cumarinici sono composti a basso peso molecolare, rapidamente e facilmente assorbiti se somministrati per via orale. Si legano alle proteine plasmatiche (albumina) per il 97-99%, cosicché soltanto una piccola frazione di tutta la sostanza (quella libera, in equilibrio dinamico con quella legata) è farmacologicamente attiva. Per il loro meccanismo di azione questi farmaci vengono denominati anti-vitamina K (AVK). La loro emivita plasmatica, e di conseguenza la loro durata di azione, variano in rapporto al tipo di farmaco e alla dose somministrata. Il metabolismo dei cumarinici avviene quasi totalmente nel fegato, mentre i loro metaboliti (in parte ancora farmacologicamente attivi) vengono escreti nelle urine e nelle feci.
Gli AVK agiscono bloccando, negli epatociti, la riduzione della vitamina K-epossido a vitamina K, mediante inibizione competitiva di specifiche epossido-reduttasi. In questo modo (poiché l‘enzima gamma-glutamilcarbossilasi necessita di vitamina K ridotta come cofattore per la sua attività) viene impedita la gamma-carbossilazione dei fattori II, VII, IX, X, già sintetizzati dalle cellule epatiche, carbossilazione che è, a sua volta, indispensabile per la loro attività biologica. È grazie infatti ai residui di acido gamma-carbossiglutammico che i fattori si legano, tramite ioni calcio, alle superfici fosfolipidiche a carica negativa delle cellule su cui avvengono le reazioni coagulative. Tale effetto è proporzionale alla dose di farmaco assunta, a parità di molte altre condizioni biologiche e cliniche. Tuttavia, allo stesso modo gli AVK inibiscono anche la funzione di due importanti inibitori naturali della coagulazione, la proteina C e la proteina S, fatto che può determinare un effetto avverso raro ma molto grave, la necrosi cutanea da warfarin (29). In Italia sono attualmente registrati due farmaci AVK: warfarin ed acenocumarolo.
Fattori genetici nella variabilità della risposta agli AVK
Sono state descritte numerose mutazioni del gene codificante per il citocromo P450 CYP2C9, responsabile del metabolismo ossidativo del più potente enantiomero S del warfarin, che ne modificano la farmacocinetica (30). Sono state poi descritte mutazioni del gene codificante per l‘enzima vitamina K epossido-reduttasi (VKORC1) che possono causare resistenza al warfarin in alcuni individui (31). La tipizzazione genetica del CYP2C9 e del VKORC1 consentirebbe di ottenere un buon modello predittivo per la stima del fabbisogno di AVK (32) nei pazienti che inizino una terapia con AVK (principalmente per ridurre casi iniziali di sovradosaggio).
Tuttavia, nella pratica clinica questo è difficile a realizzarsi (costo dei test e tempi richiesti per ottenere i risultati) e di scarsa rilevanza se il paziente viene seguito con attenzione da medici esperti nella gestione degli AVK. Di fatto, tali test non sono mai entrati nella pratica clinica e non vengono più raccomandati dalle società scientifiche (29).
Antidoto e neutralizzazione dell‘effetto degli AVK
La somministrazione di vitamina K1 determina una regressione dell‘effetto anticoagulante degli AVK più rapido della sola interruzione della loro somministrazione. La vitamina K può essere somministrata per bocca o per via parenterale, quest‘ultima con un effetto più rapido e rilevante, ma che richiede comunque alcune ore per normalizzare il valore di INR (33).
Pertanto, la sola somministrazione di vitamina K1 non risulta sufficiente per emorragie maggiori in urgenza ed emergenza. In tali condizioni risulta necessaria una terapia sostitutiva con concentrati del complesso protrombinico o, se non disponibili, con plasma fresco congelato che, tuttavia, potrebbe non correggere completamente il deficit di coagulazione, soprattutto per elevati valori di INR (34,35).
I nuovi farmaci anticoagulanti orali
Lo sviluppo di nuovi farmaci anticoagulanti orali diretti verso un target specifico (anticoagulanti orali diretti, DOAC) è una novità di grande rilevanza sia sul piano della ricerca farmacologica che su quello clinico. La sperimentazione sui nuovi DOAC è in continua espansione, sia con la proposta di nuove molecole sia con l‘impiego in trial clinici di elevata numerosità e qualità metodologica, pubblicati sulle più importanti riviste scientifiche. Contrariamente agli anticoagulanti orali fin ora disponibili (AVK), che riducono la sintesi di numerosi fattori attivi, la caratteristica saliente dei DOAC è il legame diretto con uno specifico fattore della cascata della coagulazione, che ne determina l‘inibizione.
Le molecole di DOAC oggi a disposizione (o disponibili entro breve, già ora in avanzato stadio di sperimentazione) sono rappresentate da un inibitore diretto del fattore IIa (dabigatran etexilato) e da alcuni inibitori del fattore Xa (rivaroxaban, apixaban, edoxaban, betrixaban ed altri) (36). Le idee innovative che sono alla base dello sviluppo dei DOAC, l‘elevata qualità nella ricerca farmacologica ed i cospicui investimenti nella sperimentazione clinica portano a promesse di efficacia, sicurezza ed ampia possibilità di impiego dei DOAC, promesse che hanno trovato riscontro nei risultati degli studi clinici ad oggi pubblicati (Tab. 1).

Tuttavia, è necessario che i profili farmacologici, e quindi pregi, limiti e modalità d‘impiego dei DOAC, vengano ben compresi prima del loro utilizzo. La dimostrata semplicità di uso dei DOAC in molti pazienti non deve tradursi in un eccesso di semplificazione nella scelta e confidenza nei risultati, con il rischio che poi l‘iniziale entusiasmo si trasformi in un ingiustificato timore e scetticismo.
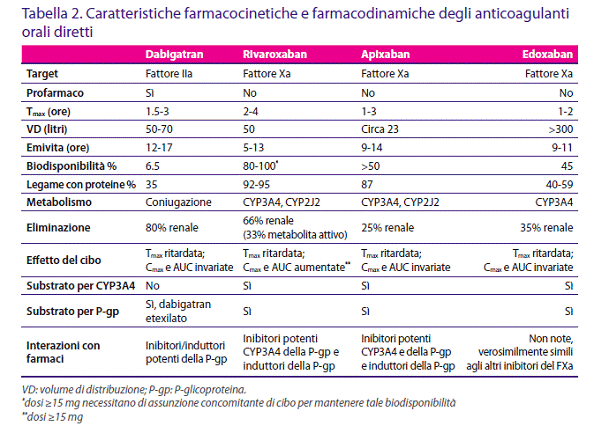
Le caratteristiche farmacocinetiche e farmacodinamiche salienti dei DOAC già disponibili, o in più avanzata fase di studio, sono riportate nella Tabella 2.
Inibitori diretti della trombina
Come noto, la trombina (FIIa) gioca un ruolo chiave nei meccanismi della coagulazione attivando la conversione del fibrinogeno in fibrina, attivando altri substrati (fattori V, VIII, XI e XIII) ed attivando recettori piastrinici mediatori dell‘aggregazione. Gli inibitori diretti della trombina si legano direttamente al sito attivo del FIIa ed hanno un effetto prevedibile sull‘inibizione della formazione del coagulo/trombo (37) (Fig. 1).
La categoria degli inibitori diretti del FIIa è attualmente rappresentata da una sola molecola (dabigatran etexilato). Tuttavia, alcuni anni or sono una molecola (melagatran/ ximelagatran) con medesimo target era già stata sviluppata ed aveva dimostrato promettente efficacia in numerosi studi clinici, anche di fase III (38,39). Ximelagatran, tuttavia, era poi stato ritirato dal commercio, dopo la sua iniziale approvazione da parte di alcuni enti regolatori, a causa di una potenziale tossicità epatica dalle conseguenze difficilmente prevedibili e prevenibili.
Dabigatran etexilato
Dabigatran etexilato è il profarmaco di dabigatran, inibitore della trombina non covalente. Dabigatran è altamente specifico per la trombina; si dissocia rapidamente da questo enzima lasciandone una piccola quota attiva per i meccanismi dell‘emostasi.
Dabigatran non è assorbibile per via orale ed è pertanto stato sviluppato un profarmaco lipofilo in grado di essere assorbito dopo somministrazione per bocca. Tale profarmaco, dabigatran etexilato, viene rapidamente convertito nel suo metabolita attivo per idrolisi da parte di esterasi nelle cellule intestinali, vena porta e fegato (40,41). Il picco plasmatico (Cmax) viene raggiunto dopo circa 2 ore dalla somministrazione. Quindi, nonostante la bassa biodisponibilità di dabigatran (6-7%) si ottiene una farmacocinetica lineare con Cmax e area sotto la curva (AUC) che aumenta proporzionalmente dopo la somministrazione di una singola dose (41).
 Per ottenere un assorbimento ottimale, dabigatran etexilato necessita di un ambiente acido. Per conseguire tale effetto, indipendentemente dal pH gastrico, la capsula elaborata è composta da uno strato di farmaco che contiene un nucleo di acido tartarico. Tale formulazione crea un microambiente acido e migliora l‘assorbimento, ma è stata messa in relazione con i problemi di dispepsia che sono stati segnalati in alcuni pazienti negli studi clinici di fase II e III (42). Da quanto detto deriva l‘importanza di mantenere l‘integrità della capsula rigida, per evitare variazioni anche rilevanti nella biodisponibilità del farmaco. Tale integrità va anche salvaguardata da problemi di umidità che renderebbero instabile la formulazione: per tale motivo il blister originario che contiene le capsule non va manomesso e le capsule non vanno estratte prima dell‘ingestione. L‘influenza dell‘ambiente acido nell‘assorbimento del farmaco è potenzialmente condizionata dalla contemporanea assunzione di antiacidi o inibitori di pompa protonica. La somministrazione di pantoprazolo, ad esempio, riduce l‘AUC di circa il 20% e la Cmax di circa il 30% (43). Pertanto, si raccomanda di somministrare dabigatran etexilato almeno 2 ore prima di un eventuale farmaco antiacido. Invece, l‘assunzione di dabigatran durante il pasto, anche se può ritardare il raggiungimento della Cmax, non interferisce con biodisponibilità, AUC o concentrazione al picco.
Per ottenere un assorbimento ottimale, dabigatran etexilato necessita di un ambiente acido. Per conseguire tale effetto, indipendentemente dal pH gastrico, la capsula elaborata è composta da uno strato di farmaco che contiene un nucleo di acido tartarico. Tale formulazione crea un microambiente acido e migliora l‘assorbimento, ma è stata messa in relazione con i problemi di dispepsia che sono stati segnalati in alcuni pazienti negli studi clinici di fase II e III (42). Da quanto detto deriva l‘importanza di mantenere l‘integrità della capsula rigida, per evitare variazioni anche rilevanti nella biodisponibilità del farmaco. Tale integrità va anche salvaguardata da problemi di umidità che renderebbero instabile la formulazione: per tale motivo il blister originario che contiene le capsule non va manomesso e le capsule non vanno estratte prima dell‘ingestione. L‘influenza dell‘ambiente acido nell‘assorbimento del farmaco è potenzialmente condizionata dalla contemporanea assunzione di antiacidi o inibitori di pompa protonica. La somministrazione di pantoprazolo, ad esempio, riduce l‘AUC di circa il 20% e la Cmax di circa il 30% (43). Pertanto, si raccomanda di somministrare dabigatran etexilato almeno 2 ore prima di un eventuale farmaco antiacido. Invece, l‘assunzione di dabigatran durante il pasto, anche se può ritardare il raggiungimento della Cmax, non interferisce con biodisponibilità, AUC o concentrazione al picco.
Dopo il raggiungimento della Cmax, la riduzione della concentrazione plasmatica avviene in modalità bifasica caratterizzata da una rapida fase di distribuzione, con riduzione a meno del 30% dell‘AUC in 4-6 ore nel giovane e in circa 12 ore nell‘anziano (44). Dabigatran ha un legame di circa il 35% con le proteine ed un volume di distribuzione (VD) di 50-70 litri, indicando una moderata distribuzione tissutale (Tab. 2).
Metabolismo
Dabigatran etexilato è metabolizzato da parte di esterasi plasmatiche che, tramite coniugazione ed idrolisi, lo convertono nella forma attiva dabigatran (45). Non è metabolizzato dal sistema del citocromo (CYP) 450 e non inibisce o induce alcuna attività del CYP.
Escrezione
La via di escrezione principale è renale (circa 80%): il farmaco rimanente viene coniugato ed escreto con la bile. Una riduzione della funzione renale prolunga l‘emivita di dabigatran (41). Pazienti con moderato deficit di funzione renale (clearance della creatinina [CrCl] 31-50 ml/min) dimostrano un incremento dell‘AUC di 3.2 volte rispetto ai controlli sani, mentre in caso di deficit grave di funzione renale l‘AUC aumenta di 6.3 volte, con incremento modesto di Cmax e tempo di picco (Tmax), ma un raddoppiamento dell‘emivita (46). Questo comporta la necessità di ridurre la dose in caso di deficit di funzione renale. Si stima che l‘emodialisi possa rimuovere circa il 65% del farmaco, ma l‘ampio VD e la distribuzione tissutale suggeriscono che siano necessari tempi di emodialisi più lunghi per una sua completa eliminazione (47). Non vi sono, invece, significative variazioni nel metabolismo del farmaco dovute all‘età, genere o moderato deficit epatico (Child-Pugh B). Tuttavia, i pazienti con grave deficit epatico sono stati esclusi dai trial e pertanto non vi sono solidi dati clinici in merito.
Un antidoto specifico per dabigatran è stato identificato ed è in sperimentazione (48), ma non è ancora disponibile per uso clinico.

Interazioni
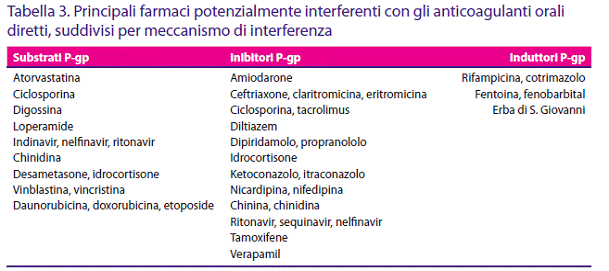
Dabigatran è un substrato per il sistema di trasporto intracellulare della P-glicoproteina (P-gp), che ha un ruolo importante nell‘assorbimento e distribuzione dei farmaci. La P-gp è espressa ampiamente nell‘organismo, sulla superficie del tratto gastrointestinale, cervello, fegato, reni e capillari: agisce come una pompa di eliminazione che previene l‘ingresso di sostanze estranee. Il controllo dell‘efficienza di tale sistema è parzialmente di tipo genetico, ma cibi e farmaci ne possono influenzare l‘attività. Farmaci che inibiscono la P-gp incrementano l‘assorbimento di un substrato, mentre farmaci che la inducono determinano l‘effetto opposto. Esistono numerosi induttori o inibitori della P-gp, tra i quali farmaci frequentemente assunti da pazienti con fibrillazione atriale. La potenziale influenza di tali interazioni farmacologiche non è trascurabile (49).
La cosomministrazione di forti induttori della P-gp come la rifampicina ha determinato una significativa riduzione sia dell‘AUC che della Cmax (65-66%). Forti inibitori della P-gp, quali il ketoconazolo, determinano un notevole incremento di AUC e Cmax (circa 150%). L‘interazione con un altro inibitore della P-gp, il verapamil, risulta dipendente dalla dose e dalla vicinanza della sua somministrazione, che si raccomanda pertanto non avvenga prima di 2 ore dall‘assunzione di dabigatran, così come per amiodarone, claritromicina o chinidina (50) (Tab. 3).
Ovviamente esiste un aumento del rischio emorragico in caso di concomitante somministrazione di altri farmaci anticoagulanti e/o antiaggreganti.
Inibitori del fattore Xa
Il fattore Xa (FXa) della coagulazione si lega al fattore Va sulla superficie delle piastrine attivate per formare il complesso protrombinasi, che converte la protrombina in trombina (fattore II → IIa). Il FXa si è dimostrato un buon target per farmaci anticoagulanti. Gli inibitori del FXa si legano selettivamente e reversibilmente al sito attivo del FXa e ne bloccano l‘interazione con il suo substrato, inibendo pertanto l‘effetto finale di generazione della trombina. Gli inibitori diretti del FXa bloccano sia il FXa libero che quello incorporato nel complesso protrombinasi (Fig. 2).

Esistono vantaggi teorici nel blocco del FXa: la generazione della trombina è inibita sia dalla via intrinseca sia da quella estrinseca, in quanto il FX è collocato all‘intersezione di queste due (51). Bloccando più a monte la cascata della coagulazione si previene l‘amplificazione della generazione della trombina e pertanto potrebbe essere richiesta una minor quantità di farmaco per ottenere l‘inibizione, se paragonata ad una inibizione diretta della trombina. Inoltre, il mantenimento di tracce di trombina attive per l‘emostasi potrebbe facilitare processi emostatici locali, riducendo gli eventi emorragici ed aumentando il profilo di sicurezza. Inoltre il FXa, contrariamente al FIIa, ha minime funzioni al di fuori dell‘attività emostatica. Gli inibitori orali del FXa hanno dimostrato un effetto anticoagulante prevedibile (52).
Sono già stati individuati antidoti per neutralizzare l‘effetto anticoagulante degli inibitori del FXa che, tuttavia, non sono ancora disponibili per uso clinico (53).
Rivaroxaban
È un inibitore selettivo e potente del FXa che non manifesta attività inibitoria su altre serinproteasi come trombina, plasmina, FVIIa, FIXa o proteina C attivata. Inibisce il FXa in modo rapido e competitivo, bloccando sia quello libero che la protrombinasi e quello all‘interno di un coagulo. Non determina effetti diretti sull‘aggregazione piastrinica (54). Vi è una buona correlazione fra la concentrazione plasmatica di rivaroxaban e l‘inibizione del FXa. L‘effetto di inibizione, dopo singola dose, viene mantenuto per 8-12 ore dopo dosi superiori a 5 mg, e non ritorna al valore iniziale prima di 24 ore.
 Tali caratteristiche cinetiche permettono di utilizzare regimi terapeutici con monosomministrazione giornaliera. La somministrazione di rivaroxaban determina un allungamento dose-dipendente di tempo di protrombina (PT) ed aPTT, con una correlazione abbastanza lineare tra la concentrazione plasmatica e questi test di coagulazione (55). È stata formulata un‘interessante proposta per la definizione e la calibrazione della specifica sensibilità delle tromboplastine utilizzate per la determinazione del PT nei confronti di plasmi calibrati con quantità note di rivaroxaban. Questa procedura potrebbe portare all‘elaborazione di un semplice sistema, simile a quello usato per i cumarinici, per la valutazione di quantità di rivaroxaban in circolo (56). È inoltre già disponibile un metodo quantitativo cromogenico per il dosaggio delle concentrazioni di rivaroxaban (57).
Tali caratteristiche cinetiche permettono di utilizzare regimi terapeutici con monosomministrazione giornaliera. La somministrazione di rivaroxaban determina un allungamento dose-dipendente di tempo di protrombina (PT) ed aPTT, con una correlazione abbastanza lineare tra la concentrazione plasmatica e questi test di coagulazione (55). È stata formulata un‘interessante proposta per la definizione e la calibrazione della specifica sensibilità delle tromboplastine utilizzate per la determinazione del PT nei confronti di plasmi calibrati con quantità note di rivaroxaban. Questa procedura potrebbe portare all‘elaborazione di un semplice sistema, simile a quello usato per i cumarinici, per la valutazione di quantità di rivaroxaban in circolo (56). È inoltre già disponibile un metodo quantitativo cromogenico per il dosaggio delle concentrazioni di rivaroxaban (57).
Assorbimento e distribuzione
Rivaroxaban è rapidamente assorbito dopo somministrazione per via orale, raggiungendo una Cmax dopo 2-4 ore (51). Dopo dosi multiple si osserva un aumento della AUC proporzionale alla dose (58). Per dosi inferiori a 10 mg la biodisponibilità è dell‘ordine di 80-100%; per dosi superiori di 15 o 20 mg in somministrazione unica si osserva una riduzione della biodisponibilità del 33%, ma tale effetto può essere revertito completamente con l‘assunzione della compressa ai pasti (incremento del 30-40% dell‘AUC e del 76% della Cmax). Pertanto tali dosi andrebbero assunte con il cibo (58). Nel plasma, rivaroxaban mostra un legame superiore al 90% con le proteine ed un basso VD. Come per dabigatran, anche rivaroxaban è un substrato per la P-gp, con alcune implicazioni nelle interazioni con farmaci.
Metabolismo
Rivaroxaban è parzialmente metabolizzato tramite il sistema CYP450, specificamente da CYP3A4/5 e CYP2J2. Pertanto, i pazienti con deficit di funzione epatica moderata (Child-Pugh B) dimostrano una clearance ridotta, con significativi incrementi dell‘AUC (59). Rivaroxaban non andrebbe quindi utilizzato nei pazienti con disfunzione epatica moderata o grave, che sono di fatto stati esclusi dagli studi.
Escrezione
Due terzi di rivaroxaban vengono rinvenuti nelle urine: il 36% di tale quantità risulta immodificato. Un terzo viene escreto per via epatobiliare con le feci (51,58). L‘emivita di 5-9 ore risulta aumentata a 11-13 ore nei pazienti anziani a causa del declino della funzione renale correlato all‘età. Pertanto, nel paziente con insufficienza renale moderata è necessaria cautela: è raccomandata una riduzione della dose per pazienti con una CrCl 15-50 ml/min e vi è controindicazione all‘uso nei pazienti con grave disfunzione renale (negli studi, con CrCl <30 ml/ min). La farmacocinetica di rivaroxaban non è sostanzialmente alterata dal genere o dal peso corporeo, anche se un incremento del 24% della Cmax si è osservato nei pazienti di peso ≤50 kg. Tuttavia, in considerazione della prevalenza del rischio trombotico rispetto a quello emorragico, le Autorità Regolatorie hanno preferito consigliare l‘uso della dose piena da 20 mg nella terapia di mantenimento di questi pazienti.
Interazioni con farmaci
Come visto, rivaroxaban è un substrato per CYP3A4 e P-gp. Pertanto, viene suggerita cautela in caso di uso di induttori di CYP3A4 e P-gp (come la rifampicina), mentre è da evitare l‘uso contemporaneo di forti inibitori (come antifungini azolici o antiretrovirali inibitori delle proteasi). La cosomministrazione di ketoconazolo ha determinato un incremento clinicamente rilevante, superiore alle 2 volte, dell‘AUC e della Cmax di rivaroxaban; lo stesso si è verificato con ritonavir. La cosomministrazione di rifampicina ha determinato una riduzione di circa il 50% della concentrazione plasmatica (43); tali farmaci andrebbero, pertanto, rispettivamente evitati o usati con cautela in corso di terapia con rivaroxaban, o viceversa (Tab. 3). Come per tutti gli anticoagulanti, è necessaria cautela nella cosomministrazione di altri farmaci anticoagulanti e/o antiaggreganti per l‘incremento del rischio emorragico.
Uno studio sulla neutralizzazione dell‘effetto di rivaroxaban ha dimostrato che la somministrazione di concentrato del complesso protrombinico ne neutralizza l‘effetto anticoagulante, valutato con test dell‘emostasi, in volontari sani (60), fornendo un presupposto biologico al trattamento delle emorragie maggiori in attesa della disponibilità di un antidoto specifico.
Apixaban
Apixaban è una molecola di piccole dimensioni con attività selettiva di inibizione del FXa con proprietà di farmacocinetica lineare e che determina un prolungamento dose-dipendente di PT ed aPTT. Come per rivaroxaban, l‘attività anti-FXa è in ottima correlazione con la concentrazione plasmatica del farmaco (61).
Assorbimento e distribuzione
Apixaban è rapidamente assorbito nello stomaco e nell‘intestino, raggiungendo una Cmax dopo 1-3 ore, con una biodisponibilità di circa 50%. Lo steady state nella concentrazione plasmatica viene raggiunto in 3 giorni. Apixaban ha un basso VD ed il suo legame con le proteine è dell‘87%. Non vi sono significative differenze in Cmax, AUC ed emivita in relazione alla contemporanea assunzione di cibo: pertanto non vi sono indicazioni ad accorgimenti di somministrazione in base ai pasti (62).
Metabolismo
Apixaban è metabolizzato principalmente tramite CYP3A4/5 e, in modo meno rilevante, da CYP1A2 e CYP2J2. Tuttavia, non si ritiene abbia effetto di inibizione o induzione diretta sul CYP450; pertanto, manifesta un basso potenziale di interferenza con altri farmaci (51,63).
Escrezione
Dopo il raggiungimento della Cmax, la concentrazione di apixaban mostra un declino iniziale rapido ed una successiva fase più lenta. Apixaban ha un‘emivita di 8-15 ore, con eliminazione tramite un doppio meccanismo: il 25% è eliminato per via renale, la parte restante per via epatobiliare nelle feci. Un deficit di funzione renale non determina effetti sulla Cmax, tuttavia l‘AUC aumenta al peggiorare della funzione renale. Una AUC del 16% in pazienti con CrCl 51-80 ml/min aumenta fino al 44% in quelli con CrCl <30 ml/min (61,64).
Sulla base di tali osservazioni e in conseguenza della limitata esperienza clinica, il farmaco viene controindicato nei pazienti in dialisi o con CrCl <30 ml/min, mentre non vi sono necessità di aggiustamento della dose nell‘insufficienza renale lieve o moderata. Apixaban è controindicato nei pazienti con coagulopatia da malattia epatica ed in genere nel paziente con grave epatopatia.
Anche se non vi sono raccomandazioni ad aggiustamento della dose, viene posta cautela nell‘uso in epatopatici Child-Pugh A o B (65). Non sono indicate variazioni delle dosi in base al peso corporeo, età, etnia o genere. Tuttavia, sono stati rilevati incrementi o diminuzioni di circa il 30% nell‘esposizione ad apixaban nei soggetti di peso corporeo rispettivamente <50 kg o >120 kg.
Inoltre, un incremento del 32% dell‘AUC si è osservato negli anziani, rispetto ad un gruppo di controllo di volontari giovani (66).
Interazioni con farmaci
In quanto metabolizzato dal sistema CYP450 e substrato per P-gp, apixaban ha potenziali interazioni con altri farmaci (61). La cosomministrazione di ketoconazolo ha causato un raddoppiamento medio dell‘AUC ed un incremento di 1.6 volte nella Cmax media (67). Farmaci che abbiano un moderato effetto inibitorio su CYP3A4 o P-gp hanno un potenziale effetto di aumentare la concentrazione di apixaban, ma in modo meno rilevante. La cosomministrazione di diltiazem, moderato inibitore di CYP3A4 e debole inibitore di P-gp, ha determinato un incremento di 1.4 volte dell‘AUC e 1.3 volte della Cmax. Risultati simili sono stati osservati con naprossene, inibitore della P-gp.
 Si suggerisce pertanto di porre attenzione alla cosomministrazione di inibitori di CYP3A4 e P-gp. Tuttavia non si ritengono necessari aggiustamenti di dose in caso di utilizzo degli inibitori meno potenti. La cosomministrazione di potenti induttori di CYP3A4 e P-gp determina una riduzione dell‘AUC e un decremento della Cmax. I dati rispettivi, relativi alla cosomministrazione di rifampicina, sono risultati -54% e -42%. Poiché tali riduzioni potrebbero determinare una diminuzione dell‘efficacia clinica di apixaban, viene raccomandata cautela nella sua somministrazione, così come nell‘uso concomitante di altri forti induttori di CYP3A4 e P-gp (Tab. 3). Come per tutti gli anticoagulanti, è necessaria cautela nella cosomministrazione di altri farmaci anticoagulanti e/o antiaggreganti, per l‘incremento del rischio emorragico.
Si suggerisce pertanto di porre attenzione alla cosomministrazione di inibitori di CYP3A4 e P-gp. Tuttavia non si ritengono necessari aggiustamenti di dose in caso di utilizzo degli inibitori meno potenti. La cosomministrazione di potenti induttori di CYP3A4 e P-gp determina una riduzione dell‘AUC e un decremento della Cmax. I dati rispettivi, relativi alla cosomministrazione di rifampicina, sono risultati -54% e -42%. Poiché tali riduzioni potrebbero determinare una diminuzione dell‘efficacia clinica di apixaban, viene raccomandata cautela nella sua somministrazione, così come nell‘uso concomitante di altri forti induttori di CYP3A4 e P-gp (Tab. 3). Come per tutti gli anticoagulanti, è necessaria cautela nella cosomministrazione di altri farmaci anticoagulanti e/o antiaggreganti, per l‘incremento del rischio emorragico.
Edoxaban
Come rivaroxaban ed apixaban, anche edoxaban è una molecola di piccole dimensioni con attività selettiva reversibile di inibizione del FXa (68). La generazione di trombina risulta inibita in modo significativo fino a 5 ore dalla somministrazione di edoxaban.
Il suo effetto antitrombotico risulta correlato alla variazione nei test di coagulazione, con il picco di inibizione del FXa 1.5 ore dopo la somministrazione, con ritorno ai valori iniziali a 12 ore. Gli studi in vitro dimostrano che edoxaban prolunga PT ed aPTT in modo concentrazione-dipendente (51). Vi è relazione proporzionale per Cmax ed AUC, indicando un profilo farmacocinetico prevedibile, con scarse variazioni intraindividuali (69).
Assorbimento e distribuzione
La concentrazione massima plasmatica dopo somministrazione orale è ottenuta ad 1-2 ore. La biodisponibilità è del 45%, mentre il VD risulta elevato per il legame relativamente scarso con le proteine: per tale motivo edoxaban potrebbe essere rimosso dalla dialisi (70). Non vi sono significative interazioni con gli alimenti e non vi sono pertanto cautele nella sua somministrazione, nei riguardi del cibo (71).
Metabolismo ed escrezione
Edoxaban viene metabolizzato principalmente tramite CYP3A4. La sua emivita risulta di 9-11 ore ed il 35% dell‘escrezione avviene per via renale.
Interazioni
Vi sono ancora scarse informazioni sulle potenziali interazioni di edoxaban con altri farmaci, anche se è ragionevole ritenerle abbastanza simili a quelle degli altri anti-FXa, in quanto anche edoxaban è un substrato per P-gp. È quindi prevedibile che i forti inibitori di P-gp ne controindichino l‘uso. In alcuni trial è stata posta l‘indicazione ad una riduzione del 50% della dose di edoxaban in caso di contemporanea somministrazione di verapamil o chinidina (72).
Bibliografia
1. McLean J. The thromboplastic action of cephalin. Am J Physiol 1916;41:250-7.
2. Andersson LO, et al. Anticoagulant properties of heparin fractionated by affinity chromatography on matrix-bound antithrombin and by gel filtration. Thromb Res 1976;9(6):575-83.
3. Lam LH, et al. The separation of active and inactive forms of heparin. Biochem Biophys Res Commun 1976;69(2):570-7.
4. Casu B, et al. The structure of heparin oligosaccharide fragments with high anti-(factor Xa) activity containing the minimal antithrombin III-binding sequence. Chemical and 13C nuclear-magnetic-resonance studies. Biochem J 1981;197(3):599-609.
5. Pini M, et al. Subcutaneous vs intravenous heparin in the treatment of deep venous thrombosis - a randomized clinical trial. Thromb Haemost 1990;64(2):222-6.
6. Hirsh J, et al. Heparin kinetics in venous thrombosis and pulmonary embolism. Circulation 1976;53(4):691-5.
7. Bjornsson TD, et al. Heparin kinetics determined by three assay methods. Clin Pharmacol Ther 1982;31(1):104-13.
8. de Swart CA, et al. Kinetics of intravenously administered heparin in normal humans. Blood 1982;60(6):1251-8.
9. Olsson P, et al. The elimination from plasma of intravenous heparin. An experimental study on dogs and humans. Acta Med Scand 1963;173:619-63.
10. Basu D, et al. A prospective study of the value of monitoring heparin treatment with the activated partial thromboplastin time. N Engl J Med 1972;287(7):324-7.
11. Kitchen S, et al. Wide variability in the sensitivity of aPTT reagents for monitoring of heparin dosage. J Clin Pathol 1996;49(1):10-4.
12. Linkins L-A, et al. Treatment and prevention of heparin-induced thrombocytopenia: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 2012;141(2) (suppl):e495S-e530S.
13. American Society of Health-system Pharmacists. Protamine Sulfate: Antiheparin Agents. Bethesda, MD: American Society of Health-system Pharmacists; 1999.
14. Harenberg J. Pharmacology of low molecular weight heparins. Semin Thromb Hemost 1990;16(suppl):12-8.
15. Bara L, et al. Pharmacokinetics of low molecular weight heparins. Acta Chir Scand Suppl 1988;543:65-72.
16. Palm M, et al. Pharmacokinetics of heparin and low molecular weight heparin fragment (Fragmin) in rabbits with impaired renal or metabolic clearance. Thromb Res 1985;40:129-33.
17. Massonnet-Castel S, et al. Partial reversal of low molecular weight heparin (PK 10169) anti-Xa activity by protamine sulfate: in vitro and in vivo study during cardiac surgery with extracorporeal circulation. Haemostasis 1986;16(2):139-46.
18. Choay J, et al. Structural studies on a biologically active hexasaccharide obtained from heparin. Ann N Y Acad Sci 1981;370:644-9.
19. Donat F, et al . The pharmacokinetics of fondaparinux sodium in healthy volunteers. Clin Pharmacokinet 2002;41(suppl 2):1-9.
20. Boneu B, et al . Pharmacokinetics and tolerance of the natural pentasaccharide (SR90107/Org31540) with high affinity to antithrombin III in man. Thromb Haemost 1995;74(6):1468-73.
21. Bijsterveld NR, et al. Ability of recombinant factor VIIa to reverse the anticoagulant effect of the pentasaccharide fondaparinux in healthy volunteers. Circulation 2002;106(20):2550-4.
22. Kuo KH, et al. Fondaparinux: a potential new therapy for HIT. Hematology 2005;10(4):271-5.
23. Dempfle CE. Minor transplacental passage of fondaparinux in vivo. N Engl J Med 2004; 350(18):1914-5.
24. Wallis RB. Hirudins: from leeches to man. Semin Thromb Hemost 1996;22(2):185-96.
25. Maraganore JM, et al. Design and characterization of hirulogs: a novel class of bivalent peptide inhibitors of thrombin. Biochemistry 1990;29(30):7095-101.
26. White HD. Pharmacological and clinical profile of bivalirudin in the treatment of patients with acute coronary syndrome. Expert Opin Drug Metab Toxicol 2009;5(5):529-38.
27. Hursting MJ, et al. Novastan (brand of argatroban): a small-molecule, direct thrombin inhibitor. Semin Thromb Hemost 1997;23(6):503-16.
28. Swan SK, et al. The pharmacokinetics and pharmacodynamics of argatroban: effects of age, gender, and hepatic or renal dysfunction. Pharmacotherapy 2000;20(3):318-29.
29. Ansell J, et al. American College of Chest Physicians. Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008;133(suppl 6):160S-198S.
30. Mannucci PM. Genetic control of anticoagulation. Lancet 1999;353:688-9.
31. Harrington DJ, et al. Pharmacodynamic resistance to warfarin associated with Val66Met substitution in vitamin K epoxide reductase complex subunit. Thromb Haemost 2005;93:23-6.
32. Sconce EA, et al. The impact of CYP2C9 and VKORC1 genetic polymorphism and patient characteristics upon warfarin dose requirements: proposal for a new dosing regimen. Blood 2005;106:2329-33.
33. Crowther MA, et al. Treatment of warfarin-associated coagulopathy with oral vitamin K: a randomised controlled trial. Lancet 2000,356:1551-3.
34. Lankiewicz MW, et al. Urgent reversal of warfarin with prothrombin complex concentrate. J Thromb Haemost 2006;4:967-97.
35. Makris M, et al. Emergency oral anticoagulant reversal: the relative efficacy of infusions of fresh frozen plasma and clotting factor concentrate on correction of the coagulopathy. Thromb Haemost 1997;77:477-80.
36. Bauer KA. New anticoagulants: Anti IIa vs. Anti Xa - is one better? J Thromb Thrombolysis 2006;21(1):67-72.
37. Di Nisio M, et al. Direct thrombin inhibitors. NEJM 2005;353(10):1028-40.
38. Executive Steering Committee on behalf of the SPORTIF III Investigators. Stroke prevention with the oral direct thrombin inhibitor ximelagatran compared with warfarin in patients with non-valvular atrial fibrillation (SPORTIF III): randomized controlled trial. Lancet 2003;362:1691-8.
39. Albers GW, et al. Ximelagatran vs warfarin for stroke prevention in patients with nonvalvular atrial fibrillation: a randomized trial. JAMA 2005;293:690-8.
40. Wienen W, et al. Effects of the direct thrombin inhibitor dabigatran and its orally active prodrug, dabigatran etexilate, on thrombus formation and bleeding time in rates. Thromb Haemost 2007;5(6):1237-42.
41. Stangier J. Clinical pharmacokinetics and pharmacodynamics of the oral direct thrombin inhibitor dabigatran etexilate. Clin Pharmacokinet 2008;47(5):285-95.
42. Connolly SJ, et al. Dabigatran versus warfarin in patients with atrial fibrillation. NEJM 2009;361(12):1139-51.
43. Nutescu E, et al. Drug and dietary interactions of warfarin and novel oral anticoagulants: an update. J Thromb Thrombolysis 2011;31:326-43.
44. Stangier J, et al. The pharmacokinetics, pharmacodynamics and tolerability of dabigatran in healthy elderly subjects. Clin Pharmacokinet 2008;47(1):47-59.
45. Blech S, et al. The metabolism and disposition of the oral direct thrombin inhibitor, dabigatran, in humans. Drug Metab Dispos 2008;36:386-99.
46. Stangier J, et al. Influence of renal impairment on the pharmacokinetics and pharmacodynamics of oral dabigatran etexilate. An open-label, parallelgroup, single-centre study. Clin Pharmacokinet 2010;49(4):259-68.
47. Chang DN, et al. Removal of dabigatran by hemodialysis. Am J Kid Dis 2013;61(3):487-9.
48. Schiele F, et al. A specific antidote for dabigatran: functional and structural characterization. Blood 2013;121(18):3554-62.
49. Lin JH, et al. Role of P-glycoprotein in pharmacokinetics: clinical implications. Clin Pharmacokinet 2003;42(1):59-98.
50. Walenga JM, et al. Drug and dietary interactions of the new and emerging oral anticoagulants. Int J Clin Pract 2010;64(7):956-67.
51. Eriksson BI, et al. Comparative pharmacodynamics and pharmacokinetics of oral direct thrombin and factor Xa inhibitors in development. Clin Pharmacokinet 2009;48:1-22.
52. Alexander JH, et al. Inhibition of factor Xa: a potential target for the development of new anticoagulants. Am J Cardiovasc Drug 2005;5(5):279-90.
53. Lu G, et al. A specific antidote for reversal of anticoagulation by direct and indirect inhibitors of coagulation factor Xa. Nature Medicine 2013;19(4):446-53.
54. Perzborn E, et al. In vitro and in vivo studies of the novel antithrombotic agent BAY 59–7939: an oral, direct factor Xa inhibitor. J Thromb Haemost 2005;3:514-21.
55. Kubitza D, et al. Safety, pharmacodynamics, and pharmacokinetics of single doses of BAY 59–7939: an oral, direct factor Xa inhibitor. Clin Pharmacol Ther 2005;78:412-21.
56. Tripodi A, et al. The International Normalized Ratio calibrated for rivaroxaban has the potential to normalize prothrombin time results for rivaroxaban-treated patients: results of an in vitro study. J Thromb Haemost 2011;9:226-8.
57. Samama MM, et al. Monitoring plasma levels of factor Xa inhibitors: how, why and when? Expert Rev Hematol 2013;6(2):155-64.
58. Kubitza D, et al. Safety, pharmacodynamics, and pharmacokinetics of BAY 59–7939: an oral, direct factor Xa inhibitor- after multiple dosing in healthy male subjects. Eur J Clin Pharmacol 2005;61:873-80.
59. Gulseth M, et al. Rivaroxaban: an oral direct inhibitor of factor Xa. Am J Health Syst Pharm 2008;65:1520-9.
60. Eerenberg ES, et al. Reversal of rivaroxaban and dabigatran by prothrombin complex concentrate. A randomized, placebo-controlled, crossover study in healthy subjects. Circulation 2011;124:1573-9.
61. Raghavan N, et al. Apixaban metabolism and pharmacokinetics after oral administration to humans. Drug Metab Dispos 2009;37(1):74-81.
62. Frost C, et al. Apixaban, an oral direct factor Xa inhibitor: multiple-dose safety, pharmacokinetics, and pharmacodynamics in healthy subjects. J Thromb Haemost 2007;5(Suppl 2):PT633. Abstract
63. Wang L, et al. In vitro assessment of metabolic drug–drug interaction potential of apixaban through cytochrome P450 phenotyping, inhibition and induction studies. Drug Metab Dispos 2010;38:448-58.
64. Ufer M. Comparative efficacy and safety of the novel oral anticoagulants dabigatran, rivaroxaban and apixaban in preclinical and clinical development. Thromb Haemost 2010;103:572-85.
65. Frost CE, et al. Single-dose safety and pharmacokinetics of apixaban in subjects with mild or moderate hepatic impairment. Clin Pharmacol Ther 2009;85(Suppl 1):S34. Abstract
66. Frost CE, et al. Effects of age and gender on the single-dose pharmacokinetics (PK) and pharmacodynamics (PD) of apixaban. J Throm Haemost 2009;7(Suppl 2):PPMO-407. Abstract
67. Frost C, et al. Effect of ketoconazole and diltiazem on the pharmacokinetics of apixaban, an oral direct factor Xa inhibitor. J Clin Pharmacol 2007;49:1091- 130. Abstract
68. Furugohri T, et al. DU-176b, a potent and orally active factor Xa inhibitor: in vitro and in vivo pharmacological profiles. J Thromb Haemost 2008;6(9):1542-9.
69. Ogata K, et al. Clinical safety, tolerability, pharmacokinetics and pharmacodynamics of the novel factor Xa inhibitor edoxaban in healthy volunteers. J Clin Pharmacol 2010;50(7):743-53.
70. Camm AJ, et al. Edoxaban: a new oral direct factor Xa inhibitor. Drugs 2011;71(12):1503-26.
71. Mendell J, et al. Effects of food on the pharmacokinetics of edoxaban, an oral direct factor Xa inhibitor, in healthy volunteers. J Clin Pharmacol 2011;51(5):687-94.
72. Ruff CT, et al. Evaluation of the novel factor Xa inhibitor edoxaban compared with warfarin in patients with atrial fibrillation: design and rationale for the Effective aNticoaGulation with factor xA next GEneration in Atrial Fibrillation-Thrombolysis In Myocardial Infarction study 48 (ENGAGE AF-TIMI 48). Am Heart J 2010;160:635-41.


-
Farmacologia clinica dei farmaci anticoagulanti
Marco Moia Centro Emofilia e Trombosi - Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico di Milano
-
Sintomi del basso apparato urinario dopo chirurgia pelvica: fisiopatologia e trattamento
Enrico Finazzi Agrò - Professore Associato; Cattedra di Urologia, Università di Roma “Tor Vergata”; UOSD Servizio di Urologia Funzionale, Policlinico Tor Vergata; IRCCS Ospedale S. Lucia, Roma
-
Fisiologia di apprendimento e memoria
Mariano Pedetti - SerT MVT, AUSL2 dell‘Umbria, Marsciano (PG)

